PROGERIA
 |
| progeria family India |

Hutchinson-Gilford Progeria Syndrome ("Progeria", or "HGPS") is a rare, fatal genetic condition characterized by an appearance of accelerated aging in children. Its name is derived from the Greek and means "prematurely old." While there are different forms of Progeria,the classic type is Hutchinson-Gilford Progeria Syndrome, which was named after the doctors who first described it in England; in 1886 by Dr.Jonathan Hutchinson and in 1897 by Dr. Hastings Gilford.
Although they are born looking healthy, children with Progeria begin to display many characteristics of accelerated aging at around 18-24 months of age. Progeria signs include growth failure, loss of body fat and hair, aged-looking skin, stiffness of joints, hip dislocation, generalized atherosclerosis, cardiovascular (heart) disease and stroke. The children have a remarkably similar appearance, despite differing ethnic backgrounds. Children with Progeria die of atherosclerosis (heart disease) at an average age of thirteen years.
Progeria affects approximately 1 in 4 - 8 million newborns. There are an estimated 200-250 children living with Progeria worldwide at any one time. It affects both sexes equally and all races. Since The Progeria Research Foundation was created in 1999, we have discovered children with Progeria living in over 40 countries.
HGPS is not usually passed down in families.
Pathophisiology:
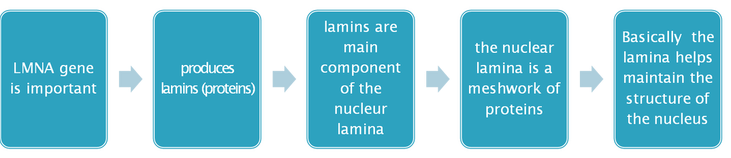
The gene defect causing HGPS and most progeroid laminopathies has been identified as a mutation in the gene LMNA, coding for the nuclear protein lamin A. Lamin A is normally expressed by most differentiated cells, and requires posttranslational farnesylation to incorporate into the nuclear membrane. The lamin A C-terminal peptide, including the farnesyl group, is subsequently cleaved, and mature lamin A becomes a prominent component of the nuclear scaffold just internal to the nuclear membrane, affecting nuclear structure and function.
In most cases, HGPS is a sporadic autosomal dominant disease caused by a single base alteration (henceforth designated as G608G) in the LMNA gene, which creates a cryptic splice site giving rise to an altered lamin A protein product in which 50 amino acids are deleted. The defective protein product in HGPS (henceforth progerin) lacks the cleavage site for removal of the C-terminal farnesylated peptide, and likely produces disease via dominant negative effects on the nuclear structure and function of various cell types that express lamin A. Most other progeroid laminopathies are caused by various mutations in the LMNA gene, which also subsequently creates abnormally functioning lamin A.


Prominent symptoms and signs of progeria are listed as follows:-
• Dwarfism / limited growth
• Alopecia or baldness
• Small face and a pinched nose
• Small jaw in comparison to head size
• Delayed tooth development
• Aged-looking, wrinkled skin
• Loss of eyebrows / hair
• Stiff joints / limited range of motion
• Frequent hip dislocations
• Premature arteriosclerosis
• Cardiovascular problems
• No sexual maturation
Their bodies are extremely fragile, like the very old,and this is because these children age at a rate that is seven times faster than that of normal children. Is it of any wonder then, that a child of ten years would give stiff competition to his seventy year old grandfather? Now the sad saga does not end with just the looks; these hapless children suffer from all the ailments of the aged and are likely to die either of heart disease, stroke or heart attack, even while in their early teens.
Case reported in India:
The family of Bisul Khan and Razia Khatooon living in Bihar India, had seven children – five of them with progeria. Three of the couple’s affected daughters are dead. Two of his progeric sons are alive, aged 23 and 22 (medical ages 70 and 66). The couple, also have two normal children.
Rural Bihar, with all its backwardness, is hardly a place for Khan’s special family. They were considered a bad omen and were ostracized.
It is not common for a family to have more than one child with progeria. Bisul Khan’s is the only family in the world with 5 cases of this dreadful condition.
A documentary titled “The 80 Year Old Children” was made in 2005, based on the real-life story of Bisul Khan’s special family
Treatment: (is at the stage of clinical trails)
The care of people with progeria is aimed at minimizing symptoms and maximizing the quality of life as much as possible.
Treatment plans are individualized and may include physical therapy occupational therapy to minimize joint stiffness and improve activity and mobility. Dietary treatments may include tube feeding for infants and supplements to help reduceweight loss.
Surgical procedures may be recommended in some children with progeria. These include pulling out the baby teeth so that prematurely erupting adult teeth have enough room to develop.
Artery bypass surgery or angioplasty may also be recommended to slow the development of cardiovascular disease.
Research:
Twenty-eight children from sixteen countries participated in the two-and-a-half year drug trial, representing 75 percent of known Progeria cases worldwide at the time the trial began. Of those, 26 are children with the classic form of Progeria.
Lonafarnib is a farnesyltransferase inhibitor that blocks the post-translational farnesylation of prelamin A and other proteins that are targets for farnesylation. Farnesylation is essential for the function of both mutant and non-mutant lamin A proteins, including progerin. Therefore, farnesyltransferase inhibitors are ideal candidates for treatment of HGPS, which is caused by a protein (progerin) that likely depends on carrying a farnesyl group to execute its aberrant functions.
Both cell culture and mouse model studies of HGPS demonstrate improved phenotype after exposure to FTI. In vitro, exposure of HGPS skin fibroblasts and progerin-transfected HeLa cells to FTIs, including lonafarnib, prevents preprogerin from intercalating into the nuclear membrane where it normally functions, and eliminates nuclear deformity. In vivo, three Progeria-like mouse models show no appreciable signs of toxicity after FTI administration. In all three of these models, disease is significantly reduced when compared to age-matched controls after oral administration of FTI.
Researchers also examined arterial stiffness (a predictor of heart attack and stroke in the general population), bone density and rigidity (indicators of osteoporosis). Every child completing the study showed improvement in an ability to gain additional weight, increased flexibility of blood vessels or improved bone structure.
Results included improvement in one or more of the following areas:
- Weight: One in three children demonstrated a greater than 50 percent increase in annual rate of weight gain or switched from weight loss to weight gain, due to increased muscle and bone mass.
- Bone Structure: On average, skeletal rigidity (which was highly abnormal at trial initiation) improved to normal levels after FTI treatment.
- Cardiovascular: Arterial stiffness, strongly associated with atherosclerosis in the general aging population, decreased by 35 percent. Vessel wall density also improved with treatment.